Enfermedad renal poliquística autosómica recesiva
Enfermedad renal poliquística autosómica recesiva: alteración hereditaria, responsable del 5 % de la enfermedades renales terminales en niños, microscópicamente hay formaciones quísticas que representan ductos dilatados, distribuidos en forma radiada tanto en corteza como médula renal, con fibrosis intersticial asociada, la ectasia puede afectar del 10 al 90 % de los túbulos colectores de lo cual depende el compromiso renal y la presentación clínica. Al mismo tiempo esta enfermedad afecta al hígado ocasionando en el fibrosis hepática congénita, como consecuencia hipertensión portal. La enfermedad renal poliquística autosómica se divide en perinatal, neonatal, infantil y juvenil, a menor edad mayor afectación. El diagnóstico es con usg en el cual los riñones se ven aumentados de tamaño, mantiene su morfología, son difusamente ecogénicos debido a las múltiples interfaces producidas por los ductos dilatados y hay perdida de la diferenciación corticomedular, a veces existe un anillo hipoecogénico periférico que corresponde a remanente de corteza comprimida, ocasionalmente se ven algunos quistes pequeños en corteza y médula, que representan los ductos ectásicos. En los estudios de seguimiento los riñones están con disminución del tamaño del riñón cambio en la ecogenicidad mayor presencia de quistes y fibrosis.
Enfermedad poliquística renal autosómica dominante: denominada como enfermedad renal poliquística del adulto, se presentan entre la 3ª y 4ª década de la vida, pudiéndose diagnosticar en la niñez de forma incidental. Generalmente los padres tienen la enfermedad, es una enfermedad multisistémica caracterizada principalmente por progresiva formación de quistes en riñones y con menos frecuencia en hígado páncreas, bazo, pulmones, ovarios y testículos, la principal causa de morbilidad es la alteración renal terminal. Al usg los riñones están agrandados, ecogénicos, con quistes y parénquima renal normal entre ellos. Los quistes son simples, mas sin embargo algunos quistes de pueden complicar con hemorragia o infección y aparecer con sedimento, septos, para lo cual se hace necesario el uso de TAC.
Enfermedad renal glomeruloquística: condición congénita rara que puede categorizarse en 3 grupos 1: hereditario asociado a síndrome de Zellweger, orofacio digital, esclerosis tuberosa trisomía 13, 2: hereditaria no sindromática y 3: forma esporádica. Histológicamente se caracteriza dilatación quística de los espacios de Bowman y dilatación variable de túbulos colectores proximales, se ha descrito igual fibrosis hepática, y dilatación de conductos biliares, la forma de presentación más habitual es masa abdominal palpable y falla renal temprana. Al ultrasonido se ve aumento de los riñones, conservando su morfología, hiperecogénicos, con pérdida de la diferenciación corticomedular y quistes periféricos pequeños, predominantemente subcasulares, aunque toda la corteza puede comprometerse.
Esclerosis tuberosa: enfermedad multisistémica heredada en forma autosómica dominante, la triada consiste en retraso mental, convulsiones y adenomas sebáceos. Los pacientes presentan hamartomas en piel, cerebro, riñones, corazón hígado, pulmones y hueso.
El hallazgo más común en el usg es múltiples quistes pequeños corticales renales y presencia de angiomiolipomas los cuales aparecen como pequeños focos ecogénicos redondeados en el parénquima los cuales igual son múltiples.
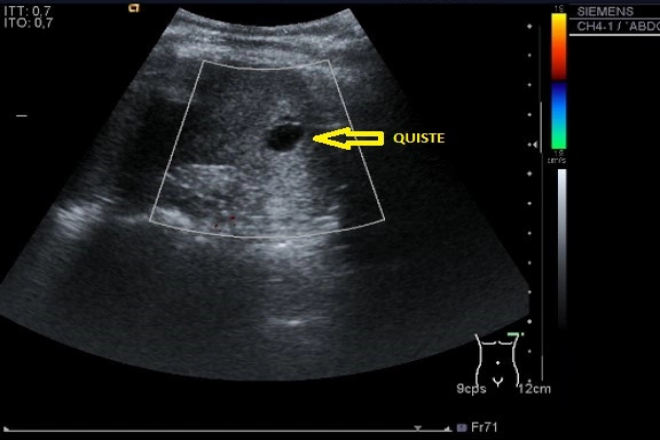
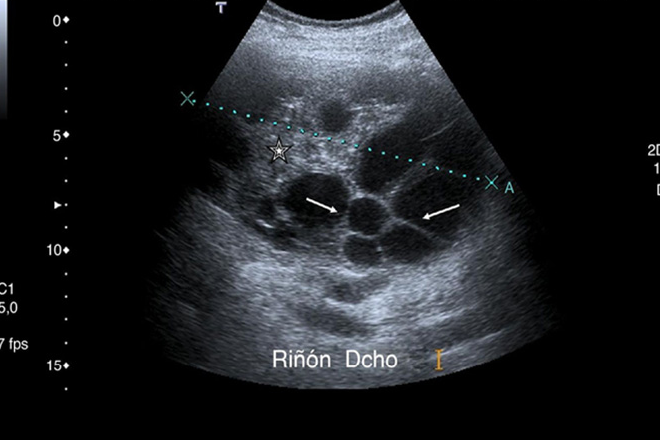
Quistes simples: son raros en niños y tienden aumentar con la edad. Generalmente asintomáticos y se diagnostican en forma incidental. Su tamaño varía de mm a varios cm, sin comunicación con el sistema colector, frecuentemente son únicos y se desarrollan en la corteza renal, y en el polo superior, raramente se complican con rotura, hemorragia o infección. Al usg estos son redondeados u oval, paredes finas o imperceptibles de contenido anecoica homogéneo, y refuerzo acústico posterior. Los quistes complejos sus paredes son gruesas, con ecos en su interior o septos internos.
Artículo escrito por el Dr. Jaime Martínez