Enfermedad de Gaucher: hallazgos por ultrasonido en el bazo
La Enfermedad de Gaucher es una afección genética hereditaria del grupo de las enfermedades lisosomales que se caracteriza por la acumulación de depósitos que el organismo no puede metabolizar, principalmente en órganos como el hígado, bazo y huesos a causa de la deficiencia de una enzima denominada glucocerebrosidasa.
La enfermedad de Gaucher es una alteración del metabolismo, poco frecuente, hereditaria autosómica recesiva, se caracteriza por acumulación de cantidades excesivas de complejos lipídicos (glucosilceramida) en las células reticuloendoteliales del bazo, hígado y medula ósea por la deficiencia de hidrolasa lisosomal B glucosidasa acida que esta codificada en el gen localizado en el cromosoma.
Más frecuente en Judíos, igualmente afectados hombres y mujeres. Se identifican 3 tipos de formas clínicas.
Tipo I.- Forma adulta no neuropatía constituye el 99% de los casos, su incidencia es de 1/1000. Son variables en su edad y presentación y esta comienza durante la infancia.
Tipo II.- Forma infantil o aguada neuropátíca, se presenta en pacientes pediátricos de 1-12 meses, la mortalidad antes de los 2 años de edad.
Tipo III.- Forma juvenil, forma Nobotien o subaguda neuropática que se presenta entre 2-6 años, sobrevida hasta la adolescencia.
Los hallazgos más importantes se encuentran en el sistema esquelético, la infiltración de la médula ósea cargada de lípidos causa pérdida de la densidad ósea con expansión y adelgazamiento en la cortical en especial el fémur. Presentan fracturas patológicas, necrosis aséptica e infartos óseos.
El bazo se encuentra aumentado de tamaño y la hepatomegalia es frecuente. Se presentan infiltrados pulmonares difusos en casos agudos.
En los laboratorios presentan fosfatasa ácida sérica, pancitopenia, leucopenia, trombocitopenia (hiperesplenismo) y hemocromatosis.
En el embarazo se puede realizar el diagnóstico mediante determinación de la actividad enzimática en las vellosidades coriónicas o en el cultivo de líquido amniótico.
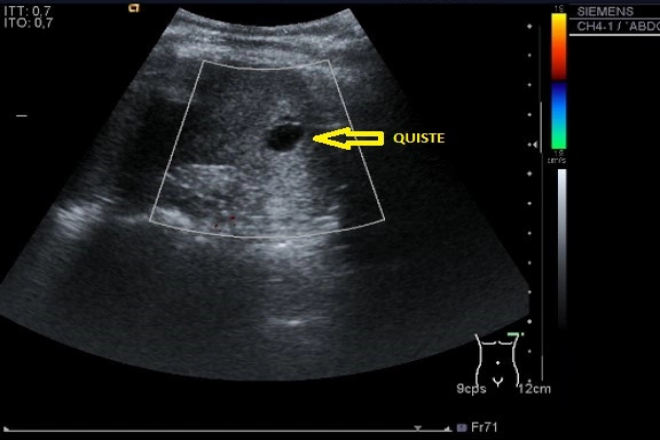
En pacientes con enfermedad de Gaucher el Ultrasonidos muestra esplenomegalia importante y en un bajo porcentaje de pacientes el bazo puede tener tamaño normal.
Las lesiones son múltiples de diferentes tamaños de 0.5 -10 cm con una media de 1.7 cm, algunos pacientes pueden presentar nódulos esplénicos simples, los cuales son hipoecogénicos bien delimitados también pueden ser hiperecogénicos irregulares con ecogenicidad mixta. También se encuentran áreas focales de contornos irregulares con ecogenicidad disminuida o aumentada, estos nódulos representan áreas focales de células de Gaucher asociados a fibrosis e infartos. A la observación existen áreas irregulares con ecogenicidad disminuida y patrón geográfico dentro del parénquima esplénico normal.
Un número mayor de pacientes presentan hepatomegalia con patrón homogéneo.
La tomografía computarizada se observan múltiples nódulos hipodensos sin reforzamiento. En la resonancia magnética se ven áreas estelares o segmentadas que son hipointensas en T1 e hiperintensas en T2 en comparación con el parénquima hepático.
El diagnóstico diferencial de lesiones de lesiones múltiples en un bazo aumentado de tamaño debe incluir enfermedad metastásicas, linfoma, infarto, absceso usualmente visto en endocarditis bacteriana subaguda, artritis reumatoide, mielodisplasia y enfermedad de Legg-Calvé-Perthes.
En cuanto a su tratamiento, si bien la enfermedad de Gaucher por ahora no tiene cura, sí se dispone de terapias que contribuyen a mejorar sus síntomas. Existen dos abordajes terapéuticos: la terapia de reemplazo enzimático, que sustituye la deficiencia de la enzima glucocerebrosidasa necesaria para la degradación de unas sustancias acumuladas en las células, y la terapia de reducción de sustrato, que reduce la producción de dichas sustancias.
Comentario.
La enfermedad de Gaucher, es una enfermedad congénita autosómica recesiva, que se caracteriza por una deficiencia de la enzima beta glucosidasa (degradación lisosómica de los glucolípidos). El gen que codifica la beta glucosidasa se localiza en el cromosoma 1 y el conocimiento del genotipo puede ser útil para valorar la severidad y valoración de la progresión de los síntomas clínicos en estos pacientes. Se manifiesta en cualquier etapa de la vida (niños, jóvenes, adultos y anciano). Este tipo de pacientes presentan hepatomegalia, esplenomegalia y osteoporosis, que afectan la calidad de vida.
La ecografía abdominal es muy importante en pacientes con la enfermedad de Gaucher, y se debe evaluar cuidadosamente del bazo, por los hallazgos patológicos encontrados, demostrables por ultrasonido como son esplenomegalia, lesiones nodulares hipoecóicas con halo de mayor ecogenidad, estos hallazgos son importantes en pacientes que presentan dolor en el cuadrante superior izquierdo.