Embarazo del Segundo trimestre, asimétria y retraso del crecimiento intrauterino.
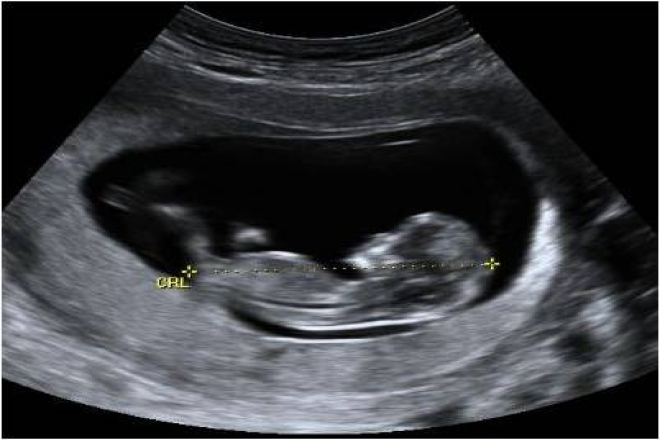
Con el fin de determinar si el feto es de crecimiento retardado, se debe evaluar las mediciones fetales contra un estándar conocido de mediciones normales para la edad menstrual ( esto no se puede hacer a menos que la edad se conoce de forma inequívoca ). La circunferencia de la cabeza de 30,9 cm está en el percentil 45 o durante 34 semanas ; la circunferencia abdominal de 25,3 cm por debajo del percentil 3 a las 34 semanas ; la longitud del fémur de 6,7 cm está en el percentil 55 en la semana 34 . El peso fetal estimado usando las tres mediciones fue 1.836 gramos, que es en el percentil 16 a las 34 semanas. La relación HC / AC fue de 1,22 que está por encima del percentil 97 en la semana 34 ; la relación FL / AC x 100 fue de 26 , lo cual está por encima del percentil 97 a las 34 semanas .
El criterio más comúnmente aceptado para el retraso del crecimiento fetal es un peso por debajo del percentil 10 para la edad. En base a esta definición, este feto no es crecimiento retardado. Sin embargo, hay otros indicadores ahora disponibles que pueden indicar retraso del crecimiento intrauterino (y por lo tanto permitir una terapia o intervención apropiada). Por ejemplo, una circunferencia abdominal por debajo del percentil 3 para la edad es una evidencia inequívoca de crecimiento fetal retardado. Además, las proporciones corporales anormales ( HC / AC , FL / CA ) indican un crecimiento abdominal retardado y caracterizan este feto de forma asimétrica crecimiento retardado. Esta es la forma más común de retraso del crecimiento intrauterino, y es generalmente el resultado de la insuficiencia placentaria en pacientes con hipertensión o diabetes de clase D mellitus. Las causas menos comunes incluyen la desnutrición severa materna, fumar en exceso, el abuso de drogas, y la anemia.
Además de los parámetros de crecimiento fetal anormal, hay otros indicadores indirectos (y menos específicas) de RCIU en este caso. Retardo del crecimiento fetal se debe sospechar cuando una placenta de grado III se identifica antes de las 36 semanas. Las primeras evidencias de la reducción del líquido amniótico (oligohidramnios) también debe sugerir la posibilidad de RCIU. Anteriormente, la medición de volumen intrauterino total se considera útil en el diagnóstico de restricción del crecimiento intrauterino, pero la complejidad del procedimiento de medida y su enfoque indirecto que sea menos útil que las mediciones fetales directos.
Se ha dicho que cuando un defecto en el sistema nervioso central significativa que está presente, también se detecta hidramnios (polihidramnios). En este caso, y señaló que en otros, es la constatación de que el líquido amniótico puede ser normal al principio de la gestación. Por lo tanto, la falta de hidramnio no cambia el diagnóstico. Niveles de alfa fetoproteína también son elevados en el líquido amniótico de la madre.
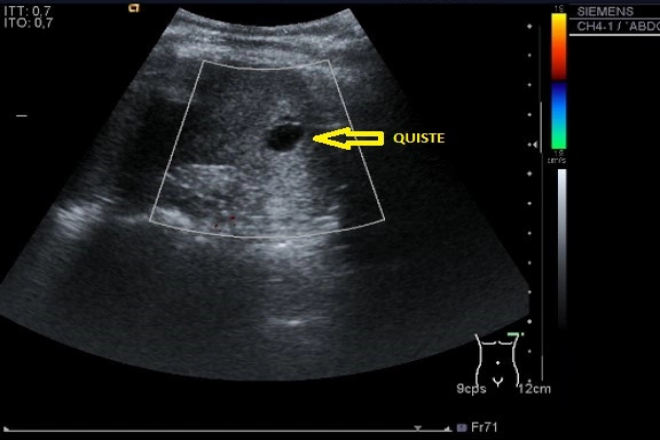
Vesícula biliar fetal normal.
Un gran número de órganos fetales normales se demuestra fácilmente con alta resolución, ultrasonido en tiempo real. Este caso demuestra la vesícula biliar fetal normal. Por lo general, de forma tubular, este cuadrante superior derecho, al parecer intrahepática, estructura quística se visualiza en aproximadamente el 50 % de los fetos. El tamaño y forma de la vesícula biliar fetal pueden ser muy variable. La ubicación de esta estructura quística ayuda a distinguirla de las venas umbilicales y portales más medial localizadas y del estómago del lado izquierdo, lleno de líquido. Los quistes de colédoco y colelitiasis también se pueden detectar en el útero.
La anencefalia .
Anencefalia es la anomalía fetal mayor más común y también es el defecto del tubo neural más común, que ocurre en de aproximadamente 1/ 1000 nacidos. Las mujeres se ven afectadas con más frecuencia que los varones con la frecuencia de 4 a 1. Factores de riesgo de anencefalia son multifactoriales e incluyen una historia familiar de defecto del tubo neural anterior.
La anencefalia es una anomalía caracterizada por la ausencia de los hemisferios cerebrales y bóveda craneal. Se cree que el resultado del fracaso de la neuroporo rostral para cerrar por 24 días de vida fetal ( 34 días menstruales ) . Aunque los hemisferios cerebrales y el cráneo están ausentes, el tronco cerebral, partes del cerebro medio, y las porciones del cráneo que forman el cartílago en la base de la bóveda craneal están presentes. El área de la cara es generalmente normal pero parece inusualmente prominente debido a la falta de el resto de la cabeza. Inicialmente , el tejido hiperecoica puede ser identificado superior a las órbitas . Fue identificado en una serie en el 45% de los casos y se denomina estroma angiomatosa (área cerebrovasculosa ) . Esto en ocasiones puede ser bastante considerable. Puede aparecer como sólidos o mixta sólida y quística. Debido a esto, hay ocasiones en las que una anomalía mucho más raro llamado exencefalia ( acrania ) puede confundirse con esta entidad .
Exencefalia se caracteriza por la ausencia total o parcial de la bóveda craneal con desarrollo completo pero anormal de tejido cerebral. Este tejido cerebral puede » desgastar » para que se vea tan anormal puede aparecer como el estoma angiomatosa . Si se identifica el tejido, la importancia de distinguir estas dos entidades es que exencefalia es una anomalía esporádica sin factores de riesgo adicionales. Sin embargo, en ambos casos, no es un resultado uniformemente fatal para el feto.
El diagnóstico diferencial incluiría también un síndrome de banda amniótica. Mientras que un síndrome de banda amniótica que implica la cabeza es un acontecimiento relativamente raro, cuando ocurre, casi siempre provoca una distorsión marcadamente asimétrica. La simetría se señala en la anencefalia y exencefalia sería, por tanto, prácticamente descartar esta entidad. Además, bandas amnióticas comúnmente afectan a otras partes del feto, en particular las extremidades.
Asociado con anencefalia es una espina bífida que se produce en el 17% de los casos , un labio leporino o paladar hendido en el 2% de los casos , y de pie en el 1,7 % de los casos . Onfaloceles también se han descrito, pero son muy poco frecuentes. Este caso tuvo ninguna otra anomalía.
El diagnóstico puede ser hecho de forma rutinaria 14 semanas de gestación. Es importante hacer con precisión, porque el resultado de estos fetos es uniformemente fatal en las primeras horas o días de vida. Incluso si no se detectan hasta que plazo, sólo el 32 % de estos fetos incluso dar lugar a nacimientos vivos. Por esta razón, la interrupción del embarazo se debe ofrecer en el momento en que se realizó el diagnóstico.
La anencefalia con estroma angiomatosa.
La anencefalia es una de las dos anomalías del sistema nervioso central más comunes, los otros meningocele o mielomeningocele siendo. Se presenta en aproximadamente 1/1000 nacimientos. Este caso representa una variante de la anencefalia donde el tejido hiperecoica mal definida se identifica superior a las órbitas. Aunque se considera poco común, una serie reciente se ha reportado su presencia en hasta un 45 % de los casos de anencefalia. Vista patológico, el tejido se corresponde con estroma angiomatosa (área cerebrovasculosa) y puede parecer bastante considerable. Esta estructura es la línea media, directamente por encima de las órbitas, y no hay bóveda craneal normales óseo se puede identificar que rodea el estroma. Estos hallazgos distinguen esta entidad a partir de ( 1 ) un encefalocele donde se esperaría una bóveda craneal a ser identificado , ( 2 ) un síndrome de banda amniótica, una condición de amputación no heredada esporádica secundaria que se produce a la interrupción amniótico y conduce a asimétrica ( no de la línea media ) deformidades, y ( 3 ) condiciones que causan la desmineralización de la bóveda craneal , tales como osteogénesis imperfecta grave. En este último, el cerebro está todavía encerrado dentro de una capa gruesa de tejido y el cerebro parece normal.
Exencefalia también debe diferenciarse de la anencefalia. Es una alteración del desarrollo extremadamente rara caracterizada por ausencia de parcial a completa de la bóveda craneal debido a una anomalía en la formación de hueso membranoso. El cerebro, mientras que el desarrollo completo, es anormal. Si las estructuras del cerebro pueden ser identificados, a continuación, esta entidad puede ser diferenciada de la anencefalia. Si no es así, los dos no pueden separarse en el útero, excepto si se encuentra de espina bífida (a veces asociado con anencefalia). Ambos son uniformemente fatal. Sin embargo, la distinción, si se puede hacer, es importante, ya que una mujer con un niño anencefálico anterior es en mayor riesgo de un defecto del tubo neural en embarazos posteriores.
Los quistes del plexo coroideo. Paciente A: feto normal. Paciente B: La trisomía 18.
Los quistes del plexo coroideo son frecuentes, lo que representa el hallazgo intracraneal más común reportado prenatalmente. Ellos son probablemente el resultado de la acumulación de líquido cefalorraquídeo y restos celulares en los pliegues epiteliales neuronales. Prácticamente todos los quistes del plexo coroideo se pueden identificar inicialmente durante el segundo trimestre y generalmente se resuelven por 24 semanas menstruales. La prevalencia de los quistes del plexo coroideo, detecta ecográficamente durante el segundo trimestre, es de aproximadamente 1 a 3%. La mayoría de los quistes del plexo choroideo son pequeños ( 3 a 10 mm de diámetro ). Con frecuencia son bilaterales, y una apariencia multilocular pueden estar presentes. Grandes quistes del plexo coroideo que llenan casi por completo el plexo coroideo pueden ampliar el ventrículo y ser confundido con la hidrocefalia.
Pequeños quistes del plexo coroideo (< 10 mm de diámetro ), sin otras anomalías fetales detectables suelen ser benignos y no dan lugar a secuelas clínicas . Demostración de pequeños quistes del plexo coroideo como un hallazgo aislado no parece justificar la amniocentesis.
Los quistes que son de gran tamaño ( > 10 mm de diámetro ), y sobre todo si son complejas, sin embargo, parecen tener un mayor riesgo de anomalía cromosómica subyacente, particularmente la trisomía 18. El riesgo específico de la trisomía 18 en un entorno de quistes del plexo coroideo en la actualidad es incierto, pero se ha sugerido para ser tan alta como 1 %. Esto es mucho mayor que la prevalencia global de la trisomía 18, que es de 1 en 6.000 embarazos. Por lo tanto, la demostración de quistes del plexo coroideo garantiza una búsqueda cuidadosa de las anormalidades adicionales que sugieren la trisomía 18 (retraso del crecimiento intrauterino, higroma quístico, macrognacia , labio leporino o paladar hendido, arteria umbilical única, o malformaciones adicionales del sistema nervioso central, corazón, tracto gastrointestinal, sistema genitourinario, o las extremidades). Detección de grandes quistes del plexo coroideo, especialmente los asociados con anormalidades detectables adicionales, parece justificar el análisis cromosómico.
La estenosis del acueducto
Numerosas anomalías intracraneales resultan en la dilatación ventricular. Normalmente, los plexos coroideos llenan los cuerpos de los ventrículos cerebrales lateral de lado a lado. Las aurículas normalmente miden menos de aproximadamente 10 mm de diámetro transversal. El tercer ventrículo normalmente se raja -como, al igual que el acueducto de Silvio y el cuarto ventrículo. La cisterna magna normalmente es de 3 a 10 mm en la profundidad anteroposterior cuando se mide en una exploración transaxial a nivel de la parte inferior del tercer ventrículo.
Aproximadamente 15 a 20% de los casos con hidrocefalia detectados prenatalmente tienen estenosis del acueducto. Puede ser adquirida o asociada con anormalidades cromosómicas. Estenosis del acueducto adquirida puede ocurrir en asociación con la infección intrauterina, hemorragia, o compresión de una masa adyacente. Estenosis del acueducto congénita puede ser ligado al cromosoma X, autosómica recesiva, o tener un patrón de herencia multifactorial. Aproximadamente el 7-27 % de los hombres con estenosis del acueducto están vinculados -X.
Las características ecográficas cardinal de la estenosis del acueducto son la dilatación de los ventrículos laterales y el tercer cuarto sin dilatación ventricular. Anomalías del sistema nervioso central extra son raros. La tasa de supervivencia global con estenosis del acueducto es de aproximadamente 90 %, pero sólo aproximadamente el 42 % de éstos tienen una inteligencia normal . Siete por ciento tienen discapacidades leves a moderadas, con un 50 % que sufren discapacidades severas. Estenosis del acueducto ligada al cromosoma X generalmente tiene un peor pronóstico, con una supervivencia de aproximadamente 80 % con prácticamente todos los sobrevivientes siendo discapacidad intelectual.
El volumen de líquido amniótico en este caso es normal. Aunque las anormalidades del sistema nervioso central significativos se asocian a menudo con polihidramnios, esto no es siempre el caso. Anomalías concomitantes conducen a oligohidramnios pueden estar presentes en algunos casos, y en otros casos la etiología exacta de la cantidad de líquido amniótico sigue siendo especulativo.
Defectos del tubo neural. Paciente 1: El mielomeningocele con hidrocefalia asociada. Paciente 2: encefalocele.
El término » defecto del tubo neural » abarca una variedad de malformaciones fetales que son la secuela de cierre anormal del eje neural fetal en desarrollo . El tubo neural está temporalmente abierto tanto craneal y caudal de 3 a 4 semanas posconcepción. El cierre del tubo neural comienza en el medio torso y procede en ambas direcciones . Ambas influencias genéticas y ambientales pueden causar malformaciones en esta etapa y resultar en la anencefalia, espina bífida, encefalocele, o la malformación de Arnold- Chiari.
Defectos del tubo neural ocurren con una incidencia de 1 en 1000 nacimientos, y cualquier niño puede tener más de una malformación. Aproximadamente el 50 % de los individuos afectados tienen anencefalia, espina bífida 50 %, y 5% encefalocele. Aunque la anencefalia es una enfermedad mortal, los defectos de la espina bífida y encefalocele son compatibles con la vida. Sin embargo, estas condiciones más comúnmente dan como resultado importante con secuelas neurológicas de larga duración morbilidad para el paciente.
La espina bífida se compone de un espectro de trastornos, de un defecto cerrado del arco neural a un saco dural abierto con mielosquisis. En el 85 % de los casos, la espina bífida es una lesión abierta (es decir, no hay piel que lo recubre), y la lesión más común y clínicamente significativa en esta categoría es el mielomeningocele. Este defecto puede ocurrir en cualquier parte a lo largo de la columna vertebral, pero es más común en la región lumbar. Defectos neurológicos van desde la anestesia menor para completar paraparesia. El ochenta por ciento de los pacientes con mielomeningocele han asociado hidrocefalia debido a una concomitante malformación de Arnold-Chiari tipo II. La base de esta malformación es una anomalía del desarrollo inferior del tronco cerebral y el cerebelo. La compresión mecánica del cuarto ventrículo por el cerebro posterior con formato incorrecto típicamente conduce a la hidrocefalia.
El encefalocele ocurre con menos frecuencia que la espina bífida (1 de cada 2.000 nacimientos). El defecto óseo que permite hernia de cualquiera de las meninges solos o el cerebro y las meninges es más común en la línea media occipital ( 75 % ) , pero puede verse en etmoidal frontal ( 13 % ) o parietal ( 12 % ) ubicaciones. La bóveda craneal fetal se puede visualizar fácilmente en detalle por el final del primer trimestre, y defectos óseos con bolsas de líquido – o – cerebrales llenado se extienden desde la bóveda craneal puede ser reconocido.
La ecografía es la más valiosa modalidad de imagen para el diagnóstico de defectos del tubo neural en el útero. La columna vertebral del feto es fácilmente evaluada por 16 a 17 semanas de desarrollo. El examen de la columna vertebral en planos sagital y axial permite la visualización de masas que surgen posterior de la columna vertebral ósea. Las imágenes axiales demuestran ensanchamiento de los elementos posteriores normalmente paralelas o convergentes en el sitio de defecto. No siempre es posible identificar con precisión los elementos neurales que entran en un mielocele, y por lo tanto la diferenciación entre un mielocele simple y mielomeningocele puede no ser siempre con confianza hizo en el útero. Ni la presencia de movimiento de las piernas del feto ni el tamaño del defecto ha demostrado ser un indicador fiable de la gravedad clínica de la malformación.
El diagnóstico serológico de los defectos del tubo neural se basa en la medición del suero materno o alfafetoproteína líquido amniótico. Este producto químico es una glicoproteína -fetal específica producida por el saco vitelino de 4 a 8 semanas de gestación, y posteriormente por el hígado fetal. Los valores normales para diferentes etapas del embarazo se han establecido, y los valores de 3 a 5 desviaciones estándar sobre el máximo normal son indicativos de defectos del tubo neural abierto. Otras anomalías que elevarán – feto- proteína alfa incluyen embarazo gemelar, muerte fetal, onfalocele, atresia intestinal alta, y la contaminación con la sangre fetal. La confirmación de un defecto del tubo neural es mediante la medición cuantitativa de la acetilcolinesterasa líquido amniótico, un producto químico no suelen estar presentes en el líquido amniótico, excepto en presencia de defectos del tubo neural abierto.
Mielomeningocele lumbosacra con malformación de Arnold -Chiari II.
Un meningocele representa protrusión de las meninges a través de un defecto de las vértebras, con la médula espinal en reposo en su posición normal dentro del canal espinal. Un mielomeningocele consta de protrusión de las meninges que contienen la médula espinal y las raíces nerviosas. Estas malformaciones son el resultado de la insuficiencia del tubo neural caudal a cerrar a las 3 a 4 semanas. El mielomeningocele es un defecto más común y grave que la meningocele y tiene una incidencia de 1 de cada 1.000 nacimientos.
Ecográficamente, mielomeningocele se puede diagnosticar cuando los centros de osificación posteriores de la columna están extendidas hacia afuera en una imagen transversal y están más separados que los centros de osificación por encima y por debajo del defecto. Una hendidura en los tejidos blandos adyacentes también suele estar presente. El saco mielomeningocele se reconoce fácilmente cuando sobresale dentro de la cavidad amniótica, como una extensión de tejido quística o blando del aspecto posterior de la columna vertebral. Sin embargo, si el saco se apoya en el miometrio o la placenta o es aplanada o intactos, la morfología anormal de los centros de osificación posteriores de la columna debe ser utilizado para hacer el diagnóstico.
Hallazgos ecográficos asociados con mielomeningocele incluyen dilatación ventricular lateral cerebral (observado en 80 a 90 % de los casos, pero no se ve en este caso), el borramiento de la cisterna magna, el «signo del banano» y el » signo del limón » (Película 4). Ventriculomegalia se define como un atrio ventricular, que mide > 10 mm. Con mayor frecuencia se asocia a la malformación de Arnold- Chiari. Las anomalías de la fosa posterior también se atribuyen a la malformación de Arnold- Chiari. El borramiento de la cisterna magna se cree que es secundaria a la compresión de la fosa posterior, con el «signo de plátano » que representa el cerebelo que envuelve el cerebro posterior tallo secundario a tracción hacia abajo de la médula espinal. El » signo del limón » se compone de un contorno frontal cóncavo o lineal de la bóveda craneal del feto, cuya etiología es incierta. Es posible que bajo presión intraespinal, que se ha observado en la mayoría de los recién nacidos con mielomeningocele, podría traducirse en el cráneo y el resultado en la deformidad hacia el interior.
Lumbosacra Meningomielocele con malformación de Arnold -Chiari tipo II.
La bóveda craneal normal es convexa en todas partes. El signo «limón» en el nivel de los cuerpos de los ventrículos laterales proporciona un poderoso pero indirecta predictor de espina bífida antes de 24 semanas de gestación (Película 3). Como un hallazgo aislado , esto puede ocurrir en el 1% de los fetos normales, pero si se suma con dilatación ventricular y / o destrucción de la cisterna magna, debe ser visto como altamente sospechosas de espina bífida.
Las vistas normales de la columna vertebral son vistas transversales, coronales y sagitales. La vista transversal es óptima para la demostración de disrafismo espinal en la que los elementos posteriores están extendidas en vez de convergente. De vez en cuando, la vista coronal muestra ensanchamiento de los elementos posteriores, pero esto puede ser muy sutil. Mielodisrafia también es bastante sutil en vistas sagital medio a menos que un gran saco mielomeningocele está presente posteriormente. De lo contrario, sutil alteración de los procesos espinales puede ser aparente. Sin embargo, las vistas longitudinales de la columna pueden indicar defectos de la segmentación de la columna vertebral que no son evidentes en las vistas transversales.
Un mielomeningocele incluye elementos de la columna vertebral en un defecto dysraphico, a diferencia de un meningocele que sólo incluye meninges. La detección de ecos lineales que se extienden en un saco que recubre un defecto dysraphico indica una fuerte evidencia de un mielomeningocele en lugar de un meningocele.
Teratoma sacrococcígeo.
Este es un caso inusual de un teratoma sacrococcígeo quística. Teratoma sacrococcígeo ocurre en aproximadamente 1 de cada 35.000 nacimientos, pero se informa, el tumor más común encontrado en los neonates. El diagnóstico prenatal de teratoma sacrococcígeo tiene una tasa muy baja de malignidad. Sin embargo, el tamaño y la extensión del tumor es más importante que la histología en los estudios prenatales. Puede ser principalmente exterior con sólo un componente presacro mínimo, predominantemente externo con un componente significativo intrapélvica, predominantemente interna con extensión intra-abdominal, o totalmente interna con ningún componente externo. La mayoría de los teratomas sacrococcígeos son sólidos o mixtos quística y sólida; sólo el 15 % son totalmente quística ( como en este caso ). Un componente intra -abdominal a menudo desplaza la vejiga urinaria por delante y puede obstruir los uréteres.
El diagnóstico diferencial depende de si la masa es predominantemente sólido o quístico y externo o interno. Otras masas sólidas o complejas en la región sacrococcígea incluyen cordoma, tumor neurogénico, lipoma, rabdomiosarcoma, hemangioma, melanoma maligno, y pseudoquiste meconio se extiende a los tejidos sólidos. Una masa predominantemente o totalmente quística pueden parecerse a los de un mielomeningocele, que debe ser excluida mediante la demostración de los elementos vertebrales posteriores intactas. Sin embargo, el teratoma puede extenderse en el canal lumbosacro y teratoma sacrococcígeo puede ocurrir con anomalías vertebrales asociadas que hacen esta distinción muy difícil. El diagnóstico diferencial de un teratoma sacrococcígeo totalmente intraabdominal incluye quiste ovárico, pseudoquiste meconial, quiste de duplicación entérica, u otras anomalías relacionadas con el intestino.
Aproximadamente el 25 % de los fetos con teratoma sacrococcígeo desarrollan hidropesía, probablemente secundaria a la vascularización del tumor. Debido al riesgo de distocia y hemorragia traumática en el tumor, el parto por cesárea se ha recomendado para los tumores mayores de 5 cm. Una masa predominantemente quística puede sufrir en la aspiración intrauterina inmediatamente antes del parto vaginal , como se ha logrado en este caso.
Unilateral displasia renal multiquística.
Enfermedad del riñón displásico Multiquístico puede ser unilateral , bilateral o segmentaria. Probablemente es secundaria a atresia del sistema ureteral yema durante la embriogénesis. El riñón afectado tiene característicamente no proximal parénquima renal normal al segmento atrésico. Aunque representa efectivamente un riñón funcional, un riñón afectado puede mostrar la formación de orina residual mínima y puede cambiar de tamaño durante la gestación.
Enfermedad del riñón displásico multiquístico unilateral lleva un buen pronóstico si el riñón contralateral es normal. Sin embargo, las anomalías de ambos riñones se han observado en el 20% de los casos en que se ha encontrado una displasia renal multiquística. La forma más severa es la enfermedad renal bilateral multicístico displásicos que tiene un pronóstico uniformemente fatal; que a menudo se asocia con otras numerosas anomalías, incluyendo sirenomelia .
Síndrome de Meckel -Gruber.
Quistes renales ocurren a menudo como una manifestación de una variedad de síndromes. Por lo tanto, la detección de quistes renales amerita una cuidadosa búsqueda de anomalías asociadas indicativos de un síndrome, sobre todo en ausencia de dilatación del tracto urinario.
En este caso, el hallazgo principal de los riñones son los riñones hiperecoicas agrandados. En el marco de oligohidramnios severo, el diagnóstico habitual que pueda ser considerada es la enfermedad renal poliquística infantil ( CIPD ) . IPCD tiene una presentación y expresividad variables, dependiendo del grado de afectación renal. La variedad perinatal produce riñones masivamente agrandados en el 90 % de los casos y la insuficiencia renal en el útero o en el nacimiento. Por lo general, la muerte ocurre en el nacimiento de la hipoplasia pulmonar. La patogénesis de este trastorno es la ectasia medular bilateral produciendo innumerables pequeñas no obstructivas de túbulos colectores renales dilatados. Las múltiples interfaces producidas por los numerosos túbulos minúsculos dan lugar a la textura renal hiperecoica característico. IPCD ocurre en aproximadamente 1:50.000 recién nacidos y se repite en el 25% de los casos como una forma autosómica dominante.
Síndrome de Meckel -Gruber es una enfermedad autosómica recesiva letal raro. Es una tríada consiste en (a ) anormalidad de los riñones, ( b ) anormalidad de la cabeza, por lo general un encefalocele; y (c ) polidactilia. Los riñones agrandados hiperecoicas asemejan a la apariencia ecográfica de IPCD, pero son una forma de displasia renal quística no obstructiva que es distinto de los riñones poliquísticos clásicos infantiles ( ectasia tubular ) y los riñones poliquísticos adultos ( grandes múltiples quistes). Las anormalidades renales ocurren en aproximadamente el 95 % de los casos con síndrome de Meckel -Gruber. Las quistes renal en el síndrome de Meckel -Gruber son muy pequeñas, casi todas por debajo de la resolución de la imagen de ultrasonido. Las múltiples interfaces causadas por los quistes microscópicos innumerables resultan en los riñones característicos hiperecoicas agrandados sin apariencia normal.
Otras causas comunes de los riñones hiperecoicas agrandados, a menudo con muy pequeños quistes microscópicos, incluyen asfixiante displasia torácica y trastornos cromosómicos.
La osteogénesis imperfecta tipo II.
El tipo letal osteogénesis imperfecta AI se produce en aproximadamente 1/60, 000 nacimientos. Es el subtipo clásico con innumerables fracturas, acortamiento de la extremidad grave ( «huesos acordeón » ) , y la disminución de la ecogenicidad ósea. Es responsable de la mayoría de los casos de osteogénesis imperfecta detectado prenatalmente. Las fracturas de costillas suelen ocurrir con este tipo de osteogénesis imperfecta. Visualización de fracturas costales prenatalmente es extremadamente rara en otros síndromes enanos. Las fracturas múltiples en forma de nervios comprimido pueden contribuir a la pequeña tórax y la hipoplasia pulmonar en este síndrome. Cráneo hipomineralización produce la visualización inusualmente clara del cerebro y puede dar lugar a la impresión errónea de la hidrocefalia.
Las fracturas costales múltiples y disminuyeron la ecogenicidad ósea son características de tipo de osteogénesis imperfecta IIA. El diagnóstico diferencial de este grave tipo de enanismo incluye hipofosfatasia, aunque hipofosfatasia característicamente tiene, huesos gráciles delgadas con moderada a severa acortamiento de las extremidades y hipomineralización difusa. Los huesos de la osteogénesis imperfecta tipo II tienden a espesar, probablemente secundaria a la formación del callo exuberante, a diferencia de los huesos delgados de hipofosfatasia.
Higroma quístico (síndrome de Turner).
Higromas quísticos son generalmente lesiones solitarias incidentales pero pueden ocurrir en el Síndrome de Turner (XO). En raras ocasiones, pueden transmitirse de forma autosómica recesiva, se pueden ver en las trisomías 13, 18, 21 y 22, y se han asociado a otros síndromes de malformaciones congénitas. La causa es una obstrucción linfática. Cuando localizada, la obstrucción linfática conduce a un higroma quístico solitario. Cuando difusa y generalizada, visto más comúnmente en el síndrome de Turner, la obstrucción también puede hacer que el líquido dentro de las cavidades del cuerpo, es decir, derrame pleural, ascitis, derrame pericárdico. En este caso , sólo con un gran quística higroma presente , la etiología no podía ser comprobada. Como resultado de ello, se realizó una amniocentesis y obteniendo cariotipo 45 XO. Esto es consistente con el síndrome de Turner. El feto murió al nacer.