Síndrome de Meckel con onfalocele y labio fisurado
El síndrome de Meckel descrito por primera vez en 1822, es una enfermedad caracterizado por la triada: riñones hiperplásicos poliquísticos (100 % de los casos), encefalocele occipital (90 %) y polidactilia postaxial (< 80 %). Este síndrome es diagnosticado usualmente mediante ultrasonido en el 2do. o 3er. trimestre del embarazo. Se reporta un caso detectado prenatalmente por ecografía obstétrica de síndrome de Meckel Gruber con onfalocele, labio fisurado y paladar hendido
PRESENTACIÓN DE CASO
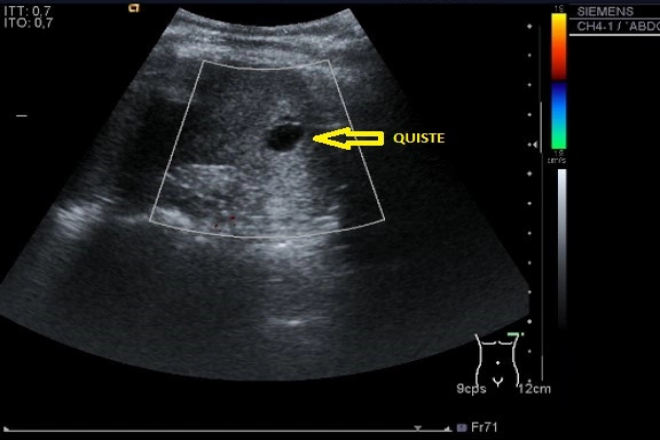
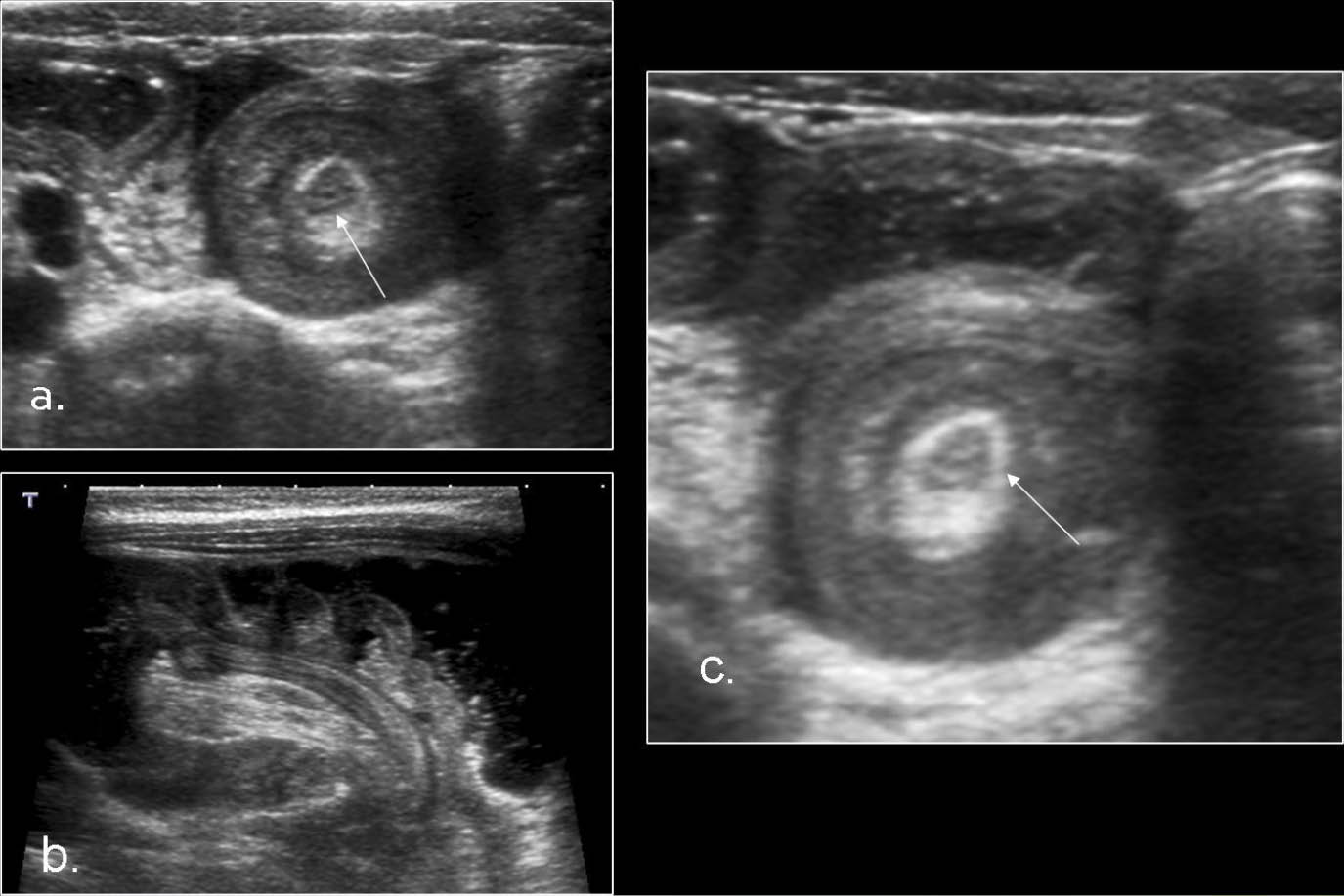
Se presenta un caso de síndrome de Meckel hijo de padres no consanguíneos, producto de embarazo de madre de 37 años, padre de 34 años, grávida 4, partos 4, a quien se le atendió el parto en un hospital de tercer nivel a las 29 semanas de gestación. Se le realizó diagnóstico prenatal al 2do. y 3er. trimestre que evidenciaba encefalocele occipital, labio-paladar hendido, polidactilia postaxial, riñones poliquísticos y canal aurículo-ventricular completo
Al examen físico se encuentra recién nacido con peso de 1 200 g, talla 39 cm, perímetro cefálico 23 cm, encefalocele occipital, labio paladar hendido bilateral, micrognatia severa, cuello corto, sinofris, polidactilia postaxial bilateral en manos, onfalocele con protrusión de hígado y asas intestinal a través del defecto con membrana íntegra, hipospadia, criptorquidia e hirsutismo en cara con Cariotipo: 46 XY. Ecocardiograma: Canal auriculo-ventricular (AV) completo.
En 1822 Johann Friedrich Meckel describió por primera vez a dos hermanos recién nacidos que murieron por malformaciones idénticas (meningoencefalocele occipital, riñones poliquísticos, polidactilia y paladar hendido). En 1934, George B. Gruber reportó varios casos similares a los que nombro «diencefalia esplacnoquística» y sugirió que la enfermedad tenía un origen genético. En 1969, Opitz y Howe propusieron el nombre de «Síndrome de Meckel».3
La prevalencia del síndrome de Meckel Gruber es mayor en Finlandia En los Estados Unidos, También se ha reportado una alta incidencia de casos entre los tártaros y los indios Gujarati.
El síndrome de Meckel es una ciliopatía causada por una mutación en un gen. Las ciliopatías incluyen además la enfermedad renal poliquística, nefronoftisis, retinitis pigmentosa, el síndrome de Bardet-Biedl, y el síndrome de Joubert.
Existen diferentes tipos de síndrome de Meckel.
Este síndrome se puede diagnosticar desde la semana 18 de gestación por ultrasonografía, o en familias consideradas de alto riesgo desde la semana 13. Aparte de las malformaciones más comunes que se presentan, que son el encefalocele occipital, polidactilia y riñones poliquísticos, también se pueden presentar hidrocefalia, agenesia de cuerpo calloso o del cerebelo, dismorfismo facial, microftalmia o anoftalmia, hipoplasia del nervio óptico, coloboma del iris, hipertelorismo, ausencia de lóbulos olfatorios, labio hendido, fisura palatina, macrostomía, lengua lobulada, micrognatia, implantación baja de orejas, cuello corto, epiglotis hendida, hipoplasia pulmonar, coartación de la aorta, esplenomegalia, asplenia, bazo accesorio, onfalocele, malrotación intestinal, ano imperforado, genitales pequeños o ambiguos, vejiga hipoplásica, malformación de Dandy-Walker, malformación de Arnold-Chiari, entre otros. El caso presentado incluye malformaciones poco frecuentes como el onfalocele, la hipospadia y el labio fisurado.
En mujeres embarazadas con fetos con síndrome de Meckel Gruber puede encontrarse un nivel elevado de a fetoproteína en sangre en el 70 % de los casos, así también los niveles de acetilcolinesterasa, gonadotropina coriónica y fosfatasa alcalina están elevados.
El pronóstico de los pacientes con este síndrome es grave, un tercio de los niños afectados muere antes de nacer, el resto sobrevive como promedio, no más de 3 h. Se menciona un caso que sobrevivió hasta los 4 meses de edad. Los diagnósticos diferenciales incluyen para este síndrome la trisomía 13, síndrome de Smith-Lemli-Opitz, síndrome C, síndrome hidroletalus, secuencia Potter, síndrome Joubert, entre otros.
El síndrome de Meckel es un desorden que se ha asociado más frecuentemente a los hijos de parejas consanguíneas. La asesoría genética incluye el cálculo de riesgo de recurrencia en las parejas que deseen tener un nuevo embarazo, el cual es estimado en el 25 % de posibilidades para un segundo embarazo con síndrome de Meckel Gruber.
El diagnóstico prenatal de riñones poliquísticos y defectos del tubo neural alerta acerca de un posible síndrome de Meckel Gruber y debe sugerir la búsqueda de malformaciones del sistema nervioso central y polidactilia.
Comentario: El síndrome de Merckel se presenta con mayor frecuencia en parejas consanguíneas cuyos hijos pueden presentar múltiples malformaciones la asociación de riñones poliquísticos, polidactilia y encefalocele, no están presentes siempre, pues la expresión puede ser muy variada como en este caso que presentaba además labio fisurado y paladar hendido, onfalocele. Debido a que este síndrome si tiene un origen genético, se debe considerar un riesgo del 25% para un nuevo embarazo.
Debido a la naturaleza de las malformaciones estas pueden ser identificadas en el periodo prenatal mediante ultrasonido lo que puede ayudar a normar el manejo de estos productos, que por su naturaleza no son compatibles con la vida ya que si llegan a término fallecen en las primeras 3 hrs de vida.
Por ello es importante tener un control ecográfico del desarrollo fetal tomando en cuenta que es un estudio de imagen accesible, relativamente barato e inocuo para el producto.